
Introduction
The (EN) ISO 13485 in its 2016 edition, as well as the FDA Guidances and the European Medical Device Regulation (MDR) require software used in the quality management systems of medical device manufacturers to be validated. While ISO 13485 and the EU MDR do not go into detail on validation should look like, the US FDA published guidelines on January 11, 2002: “General Principles of Software Validation; Final Guidance for Industry and FDA Staff”. The ISO and IEC working groups have also considered this topic and published ISO/TR 80002-2 in 2017: “Validation of software for medical device quality systems”.
The aim of this article is to demonstrate a simple way to set up tool validation.
Software List
Both ISO 13485 (in chapter 4.1.6) and the FDA Guidance (in chapter 4.8) require software validation to be commensurate with the risk of the software, regardless of the size of the company or the resources available. Accordingly, a quality management system requires an overview of the software applications used and their intended use. Preferably, this overview will also document whether the software is
- “Off the shelf” software (i.e. software that can be purchased as a mass product in stores),
- adapted software
- or even software explicitly developed for the intended use.
For standard software such as Microsoft Word, which is installed in the thousands, the probability that an error will be found through the swarm intelligence of its thousands of users is probably much higher than through tool validation. This circumstance should be taken into account in tool validation. It is possible that the process for standard software, which is only used for non-critical purposes, has already been completed at this stage.
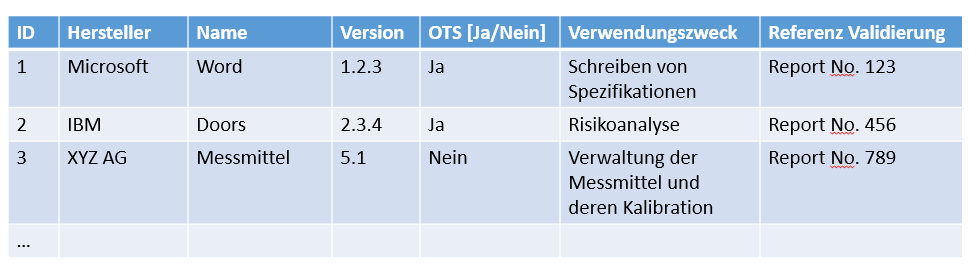
Figure 1: Software list
Software Validation Plan
The software validation plan should include the following elements:
- Description of the software including intended use, intended users and their environment
- Software requirements
- Test specifications for software requirements
- Risk management plan for the software to be validated
These elements can either all be documented in one document with different sub-chapters or they can be outsourced to different documents. The latter is particularly suitable for more extensive requirements documents.
Description of software
The chapter or document describing the software should describe the intended use, the intended users and their environment. Use case diagrams can be beneficial to give a simple, visual overview:

User requirements in the software
The user requirements should be in a testable format and given a unique ID. At IMT, we mostly use the following syntax:
| ID | Requirement |
| UR-[ID] | [Role] would like [Function] for [Purpose] |
for example, such a requirement could be
| ID | Requirement |
| UR-001 | The Serveradministrator would like to be able to reset passwords, so Users who have forgotten their password can be granted access again. |
or
| UR-002 | The User would like to save the only partially completed form, in order to be able to continue working after an interruption. |
Both requirements have a unique ID and can be tested. Requirements that cannot be tested are to be avoided. For example, “fast” should be replaced by a time such as “2 seconds”, or “bright” by “under an inspection lamp with 1500 lx”.
Test specifications for user requirements
Each user requirement requires at least one test. In addition to a unique ID, the test specification includes the test instruction and the acceptance criterion or expected result. A test for the UR-001 above could look like this:
| ID | Test steps | Expected result |
| 1 | Start Admin tool and register with a username and password with administrator rights | Admin tool starts and an Admin is logged in |
| 2 | Select the user “Vergesslich, Hans” and click on “Reset password” | Password reset window pops up |
| 3 | Enter a new password and save | New password has been set. |
| 4 | Log into the app as “Vergesslich, Hans” and enter the new password | Login successful. |
Risk management plan for this software
Since the effects of a software error in the quality management system are different from those of a medical device, the risk management plan must also be adapted accordingly. In particular, the definitions of the impact categories must be considered differently. Depending on the area of application, the same impact categories take on completely different meanings. The impact category “critical” potentially means the death of a patient in the case of a medical device, but the loss of data in the case of medical device tracking software.
Testing and assessment
Before the validation report can be produced, the software must be tested and the risks assessed.
Testbericht
The software must be tested according to the test specification. In the process, not only a “pass/fail” must be logged for each test step, but also the actual behavior observed, so that the test can be re-enacted. Therefore, the software and its environmental conditions must also be logged. This includes:
- Name and manufacturer of the software
- Version, build number
- Operating system
- 32/64-Bit environment
- Peripherals used
- ….
Date, examiner and 4-eye-principle evaluation
Using the above example, the report could look as follows:
| Test report
Environment used:
The overall test is: ☒ Passed ☐ Failed
Date of assessment: January 01, 2000
Auditor: Max Muster Evaluation: Maximilia Meier [Signature] [Signature] |
Risk analysis
The risk analysis should assess all known problems. In the case of software that is distributed for use in a regulated environment, a corresponding list of known anomalies is often also published. It is worthwhile to check the manufacturer’s support website or to contact support. In addition to problems known to the manufacturer, shortcomings such as missing features must also be assessed. If a risk is not acceptable, control measures must be defined to reduce the risk. As medical device manufacturers who are used to risk management according to ISO 14971, we recommend following the same process.
Software validation report
The software validation report should include the following elements:
- Identify software and environment
- Test report(s) or reference(s) thereto
- Risk management report on the known anomalies of the software
- Evidence that all requirements have been tested. This is most easily mapped via a traceability matrix.
- Formal evaluation and approval of the software for use
NB: For “smaller” applications, it is possible to produce the test report, risk analysis and validation report in one document.
Identifying the software and environment, test report
Often there is a matrix of validated versions and environments for the software. If this is the case, it should be mapped accordingly in the validation report:
| Software Version | Windows 10, 32 bit | Windows 10, 64 bit |
| V1.0.1, Build 1.0.1.00123 | n/a | Bericht 1.pdf |
| V1.0.1, Build 1.0.2.00254 | Bericht 2.pdf | Bericht 3.pdf |
Risk management report on the known anomalies of the software
The validation report shall further assess the residual risk arising from the use of this software. The focus is on the acceptability of the known anomalies and/or missing functions.
Traceability
Another aspect to cover is to prove that all requirements for the software have been tested. Depending on the scope of the requirements and the documentation of the tests, two ways of documenting this have emerged:
Forward linking of the requirements to the tests
A column is added to the requirement table where the requirement is checked:
| ID | Requirement | Tested in |
| UR-001 | The Serveradministrator would like to be able to reset passwords, so Users who have forgotten their password can be granted access again. | TC 1-4 |
| UR-002 | The User would like to save the only partially completed form in order to be able to continue working after an interruption. | TC 5-8 |
This variant is particularly suitable for a small scope of requirements and if the entire validation is created in one document.
Traceability Matrix
In the case of extensive requirements documents or if the test implementation has its own IDs, it is advisable to create a traceability matrix that contrasts the requirements with the test specification and possibly the test report:
| Requirement | Test specification |
Test report |
| UR-001 | TC-1 | TR-12 |
| TC-2 | TR-13 | |
| TC-3 | TR-14 | |
| TC-4 | TR-15 | |
| UR-002 | TC-5 | TR-17 |
| TC-6 | TR-18 | |
| TC-7 | TR-19 | |
| TC-8 | TR-20 |
If several reports need to be mapped for different runtime environments and software versions, this can be mapped very easily in a traceability matrix by adding additional columns.
Formal evaluation and approval
Last but not least, the entire package of documentation should be evaluated and documented with an approval decision.
Summary
As shown in the images, the validation of the software in the quality management system is nothing more than the systematic, documented execution of the upper left and right corners of the V-model widely used in software development:

This includes:
- Intended benefits and requirements
- Risk management
- Validation of user requirements
- Assessing the risks
- Assessing the overall validation and approval of the software for its intended use.
